|
Asma Brônquica
Resposta Tardia da Asma
FATORES DE TRANSCRIÇÃO
Como exposto sucintamente nos tópicos precedentes, relatamos que a patogênese da asma envolve a expressão de uma ampla série de proteínas inflamatórias, que incluem citocinas, quimiocinas, enzimas que produzem mediadores inflamatórios, receptores para mediadores inflamatórios e adesão de moléculas. O aumento da expressão da maioria destas proteínas é o resultado de um aumento na transcrição genética. Vários destes genes não são expressos em células normais sob condições habituais, porém têm sua produção aumentada por um mecanismo celular específico, nas doenças inflamatórias. O padrão de expressão da citocina determina em grande parte a natureza e a persistência da resposta inflamatória.1
A produção de RNA mensageiro (mRNA) a partir do ácido desoxirribonucleico (DNA) por uma célula é um procedimento meticulosamente regulado. O controle transcricional é o resultado de uma ação mútua entre sequências regulatórias de DNA (promotores, estimuladores e silenciadores) e as sequências específicas que se ligam a proteínas-DNA, que recebem a denominação de fatores de transcrição. Os fatores de transcrição atuam então como mensageiros nucleares que transferem informação da superfície da célula e do citoplasma para o núcleo.
Fatores de transcrição são proteínas que se ligam a sequências promotoras e regulam a transcrição gênica. A interação entre a proteína e o DNA pode resultar em indução ou repressão do gene. |
|
As mudanças na transcrição de genes são reguladas pelos chamados fatores de transcrição, que são proteínas intracelulares que se ligam a  sequências gênicas reguladoras, geralmente na 5' upstream da região promotora de genes alvo, para aumentar (transativação) e às vezes diminuir (transrepressão) a taxa de gene transcrição, resultando em aumento ou redução na síntese de citocinas, mRNA, proteínas e subsequente alteração da função celular. O excesso de ativação dos fatores de transcrição pode ser responsável por prolongada liberação de citocinas inflamatórias na asma e em certos indivíduos representa deficiência molecular primária. sequências gênicas reguladoras, geralmente na 5' upstream da região promotora de genes alvo, para aumentar (transativação) e às vezes diminuir (transrepressão) a taxa de gene transcrição, resultando em aumento ou redução na síntese de citocinas, mRNA, proteínas e subsequente alteração da função celular. O excesso de ativação dos fatores de transcrição pode ser responsável por prolongada liberação de citocinas inflamatórias na asma e em certos indivíduos representa deficiência molecular primária.
Muitos fatores de transcrição são comuns a vários tipos de células (ubíquos) e podem atuar na regulação de genes inflamatórios, enquanto que outros são específicos para certas células, podendo determinar as suas características fenotípicas.
A ativação do fator de transcrição é complexa e pode envolver múltiplas vias de transdução de sinais intracelulares, incluindo quinases [como mitogen-activated protein kinases (MAPKs), Janus kinases (JAKs) e proteinoquinase C (PKC)] estimuladas por receptores da superfície celular. A ativação das vias MAPK pelo estímulo inflamatório conduz à ativação de um número de fatores de transcrição ubíquos, como ElJ-1, c-Myc, c-Jun, c-Fos, SRF e C-enhancer-binding protein ß (C/EBPß), anteriormente denominado de NF-IL6.2 Os fatores de transcrição podem também ser ativados diretamente por ligantes (p. ex. corticoides) ou serem ativados dentro do citoplasma, expondo os sinais de localização nuclear, translocando-se para o núcleo. Os fatores de transcrição podem converter, portanto, sinais transitórios do meio ambiente junto à superfície da célula em mudanças a longo prazo na transcrição de genes, atuando assim como mensageiros nucleares (Figura 1).
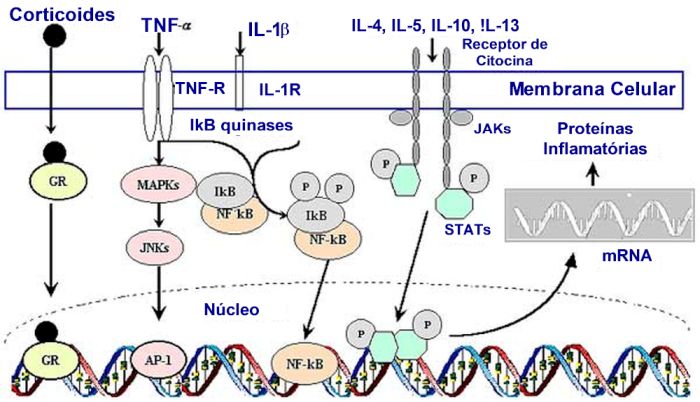
A união de fatores de transcrição aos seus motivos específicos de ligação na região promotor na dupla-hélice do DNA pode alterar a transcrição pela interação direta com os componentes da maquinaria de transcrição basal ou via cofatores que ligam o fator de transcrição à maquinaria basal de transcrição. Grandes proteínas que se ligam à maquinaria de transcrição podem ligar-se a vários fatores de transcrição e assim atuar com integradores da gene transcrição. Essas moléculas coativadoras incluem CREB-binding protein (CBP) e o p300, permitindo assim interações complexas entre diferentes vias de sinalização.3
O DNA é o responsável pelo armazenamento e transmissão da informação genética. É encontrado principalmente nos cromossomos e, em pequenas quantidades, nas mitocôndrias. O DNA de todos os cromossomos encontra-se "empacotado" em estrutura compacta, com o auxílio de proteínas básicas especializadas. Estas proteínas que se ligam ao DNA são divididas em duas classes gerais: as histonas (proteínas singulares para eucariotos) e as proteínas cromossômicas não histônicas. O complexo formado pela interação de proteínas de ambas as classes com o DNA nuclear é conhecido como cromatina (o material do qual os cromossomos são feitos). As histonas estão presentes numa quantidade tão grande (em torno de 60 milhões de moléculas de cada tipo de histona por célula) que a sua massa total de cromatina é aproximadamente igual àquela do DNA. As histonas têm um papel fundamental na compactação, de uma maneira organizada, das moléculas de DNA muito longas, em um núcleo que possui apenas uns poucos micrômeros de diâmetro.
Existem cinco tipos de histonas em dois grupos principais: as histonas nucleossômicas e os da H1. As histonas nucleossômicas são pequenas proteínas (102–135 aminoácidos) responsáveis pelo enrolamento do DNA nos nucleossomos. O nucleossomo (subunidade fundamental da cromatina) é formado por um núcleo octamérico de histonas, em volta do qual o DNA é enrolado duas vezes. A facilidade com que um segmento de DNA curva-se para dar essas duas voltas em torno de um nucleossomo varia em função da sua sequência nucleotídica.
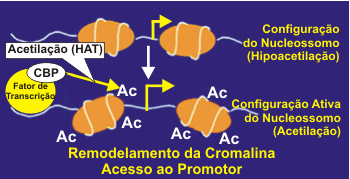 A cromatina quando visualizada ao microscópio pode apresentar-se condensada ou opaca devido ao enrolamento ou não do DNA ao redor do núcleo da histona.4 A acetilação das histonas permite o desenrolar da estrutura local do DNA, remodelando a cromatina, liberando o acesso para a ação de fatores de transcrição na região do promotor. A acetilação é regulada por um balanço entre a atividade das acetiltransferases das histonas (HATs) e histona deacetilases (HDACs) (Figura 2). A cromatina quando visualizada ao microscópio pode apresentar-se condensada ou opaca devido ao enrolamento ou não do DNA ao redor do núcleo da histona.4 A acetilação das histonas permite o desenrolar da estrutura local do DNA, remodelando a cromatina, liberando o acesso para a ação de fatores de transcrição na região do promotor. A acetilação é regulada por um balanço entre a atividade das acetiltransferases das histonas (HATs) e histona deacetilases (HDACs) (Figura 2).
A deacetilação da histona aumenta o enrolamento do DNA ao redor dos resíduos de histona, resultando em cromatina mais densa, o que determina um acesso restrito aos fatores de transcrição aos seus sítios de ligação, e desse modo reprimindo a gene transcrição.5
O papel central da CBP e proteínas associadas no controle da resposta inflamatória pode explicar o sinergismo e a repressão entre os fatores de transcrição.
Assim como os receptores de corticoides, numerosos fatores de transcrição pró-inflamatórios interagem e ativam a CRP-associated HAT activity, incluindo CREB, AP-1 (activator protein-1), NF-kB (nuclear factor kappa B), SV40 promoter-1 (SP1), E26 (Ets), NF-AT (nuclear factor of activated T cells) e STATs (signal transduction-activated transcription).6,7
O CBP e o p300 têm múltiplos domínios de ativação e podem funcionalmente interagir com vários fatores de transcrição o que pode ser a razão para explicar a ação sinérgica, por exemplo, do NF-kB e AP-1. Desde que a expressão CBP pode ser limitada na célula, pode ocorrer a competição entre fatores de transcrição pró-inflamatórios e anti-inflamatórios para quantidades limitadas de CBP, resultando na redução na expressão de genes anti-inflamatórios. A gene expressão completa depende do tipo de célula, provavelmente requer um número de fatores de transcrição agindo em conjunto, de forma coordenada, e a repressão de um único fator de transcrição sozinho pode apenas modificar parcialmente a resposta completa.
Os fatores de transcrição, embora tenham estruturas primárias diversas, formam uma estrutura terciária que é compatível com a sequência do DNA com a qual devem interagir. A grande afinidade dos fatores de transcrição para sítios específicos determina sua especificidade de ligação. Após um contato inicial feito entre a proteína e o DNA, estas interações são intensificadas devido à formação de numerosas pontes de hidrogênio, interações iônicas e hidrofóbicas. Segue-se a formação de complexos tridimensionais nucleoproteicos altamente específicos, envolvendo DNA-proteína e interações proteína-proteína. A eficiência transcricional destes complexos pode ser alterada por sutis mudanças em posições relativas ou orientações de proteínas no interior do complexo. A formação de complexos tridimensionais nucleoproteicos, altamente específicos, é frequentemente acompanhada por mudanças na conformação da proteína e no DNA, e vários fatores de transcrição induzem o DNA a se curvar e submeter-se a ligações específicas.
Os fatores de transcrição importantes na patogênese das doenças alérgicas são: activating protein-1 (AP-1), nuclear factor-kappaβ (NF-kB), nuclear factor of activated T cells (NF-AT), signal transducers and activators of transcription (STATs), guanine-adenine and thymine-adenine repeats (GATA), e o complexo corticoide receptor de glicocorticoide (GR).
Fatores de Transcrição NF-kB, AP-1, STATs e GATA 3...
As citocinas produzem seus efeitos celulares pela ativação de vários fatores de transcrição como o AP-1, o NF-kB e o STAT, os quais ativam ou reprimem os genes alvo. Estes fatores podem também intensificar e perpetuar a expressão de citocinas, visto que as regiões promotoras de muitas citocinas e seus genes de receptores revelam numerosos sítios reguladores para estes fatores de transcrição. Desde que as citocinas não atuam sozinhas, sendo produzidas e liberadas em rede bem coordenada, os níveis relativos destes fatores de transcrição podem ser responsáveis pela ação inflamatória prolongada das citocinas e seus ativadores.
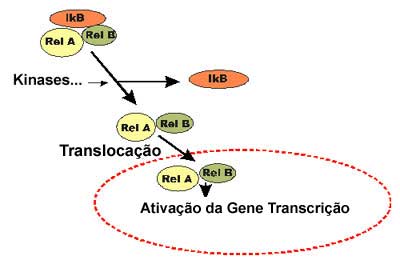
Moléculas de NF-kB existem no citoplasma da maioria das células, em forma inerte, sem ligação-DNA. O NF-kB homo e heterodímero são retidos no citosol por moléculas inibidoras, IkBα, mascarando os sinais de localização (NLS) das proteínas NF-kB e mantendo-as sequestradas em estado inativo no citoplasma. Existem várias proteínas NF-kB diferentes. A forma clássica é um heterodímero, uma proteína (p65) de 65-kDa (Rel A) e uma subunidade (p50) de 50-kDa (Rel B). A ativação extracelular por vírus (rinovírus), oxidantes (NO, O3), citocinas (TNF-α, IL-1ß...), lipopolissacarídeo (LPS) etc determina a ativação da IkB quinase através de uma via ainda mal definida. A ativação do NF-kB ocorre predominantemente por fosforilação e degradação das proteínas IkB. Após a fosforilação das proteínas IkB segue-se a poliubiquitinização e elas são degradadas como parte de seus respectivos complexos ternários. O NF-kB livre transloca-se para o núcleo, onde upregulate a expressão de múltiplos genes envolvidos nas respostas imunes e inflamatórias, incluindo moléculas de adesão como a ICAM-1 e VCAM-1, enzimas como a COX-2 e a iNOS e a maior parte das citocinas como IL-1ß, TNF-α, IL-6, GM-CSF e quimocinas como a IL-8, RANTES, MCP-1, MIP-1α e Eotaxina.8-11 (Figuras 3 e 4) O NF-kB é também importante na ativação de células T, onde está envolvido na upregulation da IL-2 e receptores IL-2.12 O NF-kB nas células epiteliais pode apresentar grande importância ampliando a inflamação em várias doenças inflamatórias, inclusive na asma.13
No núcleo o NF-kB liga-se ao DNA nos promotores de genes alvos como um dímero usualmente composto por dois membros da família de proteínas Rel, o Rel A e o Rel B. No heterodímero NF-kB, ambas as subunidades entram em contato com o DNA, porém apenas Rel A contém o domínio de transativação que ativa a transcrição por interação direta com a maquinaria basal de transcrição.14 Na subunidade Rel B falta este domínio de transativação.
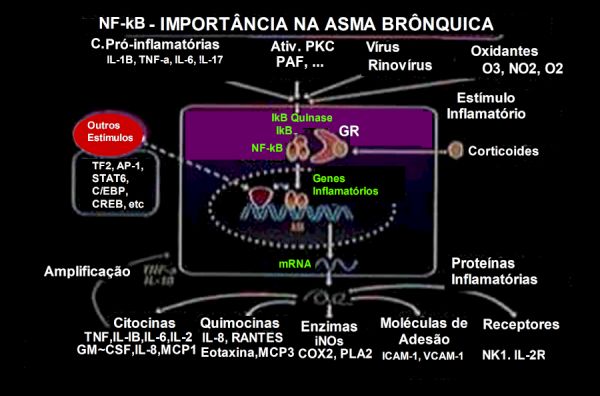
Figura 3 – A ativação de genes inflamatórios está sob controle dos fatores de transcrição como o NF-kB e AF-1. O NF-kB é particularmente importante para regular a expressão de muitos genes que estão induzidos na asma e pode apresentar um papel importante na amplificação do processo inflamatório. Por conseguinte, o efeito inibitório dos corticoides sobre o NF-kB resulta na expressão reduzida de múltiplos genes que estão ativados na asma. Figura adaptada de PJ Barnes.
Evidências sugerem uma participação central do NF-kB na patogênese da asma:
∎ O NF-kB ativado tem sido identificado em pontos-chave das vias aéreas de pacientes asmáticos.
∎ Agentes associados às exacerbações da asma como alérgenos, ozônio e infecções virais, estimulam o NF-kB. Alérgenos (p. ex. Der p 1) ativam in vitro o NF-kB em células epiteliais brônquicas15 e a exposição ao aerossol de ovoalbumina resulta em profunda ativação do NF-kB e expressão da iNOS em vias aéreas de ratos Brown Norway sensibilizados.16 Rinovírus ativam o NF-kB e aumentam a expressão genética de ICAM-1 nas células epiteliais brônquicas.17
∎ O importante papel do NF-kB tem sido demonstrado em ratos Knock-out p50(-/-). Estes animais não produzem a IL-5, citocina do tipo TH2, nem a quimiocina eotaxina, sendo desprovidos de uma resposta alérgica inflamatória eosinofílica.18
∎ O NF-κB regula a expressão de genes envolvidos na produção de muco, como os genes da mucina. O aumento da produção de muco é uma característica comum da asma e contribui para a obstrução das vias aéreas. Estudos têm demonstrado que o NF-kB desempenha um papel importante tanto na produção de muco como na regulação da expressão de MUC5B no epitélio das vias aéreas dos pacientes.19 Além disso, detectou-se que o NF-kB contribui para a fibrose peribrônquica induzida por alérgenos.20
∎ A ativação da sinalização do NF-kB é mais pronunciada em indivíduos asmáticos quando comparada a pessoas saudáveis. Prolonga o tempo de vida das células inflamatórias, que pode ser um fator contribuinte para a manutenção destas nas vias respiratórias, sustentando assim o estado inflamatório crônico.21,22 Esta via é ativada em células epiteliais brônquicas após a infecção por rinovírus, intensificando as crises de asma.23
∎ Embora pleiotrópico na ação, o maior mecanismo de ação dos corticoides, principal droga no tratamento da asma, é a inibição da ativação do NF-kB. O receptor de corticoide (GR) interage com o NF-kB pela ligação à subunidade p65 do heterodímero NF-kB,13 impedindo a ligação DNA-NF-kB,24,25 suprimindo a transcrição de genes de citocinas, quimiocinas, receptores, enzimas e moléculas de adesão (Tabela 1). Por outro lado, os corticoides podem aumentar a expressão de genes anti-inflamatórios, tais como o SLPI (secretory leukocyte protease inhibitor) e o Lipocortin-1.
Tabela 1 – Regulação da Gene Expressão pelo NF-kB
Regulação da Gene Expressão pelo NF-kB
|
| Enzimas Inflamatórias |
iNOS, 5LO, COX-2, cPLA |
| Citocinas Pró-inflamatórias |
TNF-a, IL-1ß, IL-2, IL-6, GM-CSF, G-CSF, M-CSF |
| Quimiocinas |
IL-8, MIP-1a, MCP-1, Gro-a, -ß, Eotaxina, RANTES |
| Moléculas de Adesão |
ICAM-1, VCAM-1, E-selectina |
| Receptores |
IL-2R (cadeia a), Receptor de célula T (cadeia ß) |
O AP-1 é predominantemente um heterodímero Fos/Jun que é ativado por várias citocinas, como o TNF-a e a IL-1ß, via tirosino-quinases e MAPKs.2 O c-fos, um proto-oncogene e constituinte da proteína ativadora transcripcional AP-1, está com sua expressão aumentada no epitélio de pacientes com asma.26 Em pacientes com asma esteroide resistente (AER) tem sido descrito o aumento da expressão do c-fos nas células mononucleares sanguíneas circulantes.27 O AP-1, como o NF-kB, regula muitos do genes imunes e inflamatórios que apresentam expressão aumentada na asma. A máxima ativação destes genes requer frequentemente a ativação simultânea de ambos os fatores de transcrição. Como para o NF-kB, existem fortes evidências para o aumento da expressão do c-fos no epitélio das vias aéreas de pacientes com asma,28 influenciando a regulação da expressão de genes das citocinas, participando na regulação da função celular a longo termo, no crescimento e diferenciação.
A via de sinalização Janus quinase (JAK)/STAT (signal transduction-activated transcription) é uma das vias importantes de sinalização de citocinas. A família STAT desempenha considerável participação na via de sinalização JAK/STAT, destacando-se como um componente essencial no cenário da asma e seu desenvolvimento.29
Os membros da família JAK, que são proteínas tirosina quinase não receptoras, apresentam um peso molecular aproximado de 120–140 kDa. Até o momento, identificamos quatro membros na família de proteínas JAK: JAK1, JAK2, JAK3 e TYK2. JAK1, JAK2 e TYK2 estão distribuídos em praticamente todas as células e tecidos, enquanto JAK3 é predominantemente encontrado no sistema linfático e na medula óssea.30
Por outro lado, a família STAT constitui uma classe de proteínas que atuam como alvos downstream dos JAKs. Identificamos seis membros na família STAT, nomeadamente STAT1 a STAT6. Essa interconexão entre os membros da família JAK e STAT destaca a complexidade e a importância dessa via de sinalização na regulação de uma variedade de processos biológicos fundamentais.
A supressão do gene STAT1 em células epiteliais das vias aéreas de camundongos asmáticos demonstra impacto significativo, resultando na diminuição do número de eosinófilos e na redução dos níveis de IL-5 no tecido pulmonar desses animais. Esse efeito direto contribui para a atenuação da inflamação nas vias aéreas, sugerindo uma possível abordagem terapêutica centrada na regulação da família STAT para controlar a inflamação associada à asma.31
De maneira análoga, em pesquisas envolvendo animais, a inibição seletiva do STAT3 emerge como uma estratégia eficaz na significativa redução da gravidade da inflamação pulmonar, apontando para seu potencial como alvo terapêutico promissor. O STAT3 desempenha um importante papel na modulação das células TH2/TH17 e macrófagos M2, além de estar envolvido na disfunção de células endoteliais. Essa complexa interação culmina no surgimento de sintomas associados à asma, incluindo a infiltração de eosinófilos e neutrófilos nos pulmões, hiper-responsividade brônquica e remodelação. A abordagem direcionada ao STAT3 destaca-se como uma possível estratégia terapêutica para modular esses eventos patológicos e minorar os efeitos adversos associados à inflamação pulmonar.32
O STAT-6 é um membro da família de fatores de transcrição JAK/STAT que é ativada pela IL-4. Citocinas como o GM-CSF, um potente inibidor da apoptose dos eosinófilos, e a IL-5, determinante primário da diferenciação, recrutamento, ativação, adesão e sobrevida dos eosinófilos encontram-se com níveis elevados nos locais da inflamação alérgica nas vias aéreas de pacientes asmáticos. Estas citocinas exercem primariamente seus efeitos celulares via JAK-STAT.33,34 Ratos transgênicos Knockout deficientes em STAT-6 após sensibilização alérgica não apresentam resposta a IL-4, não desenvolvem células T em resposta a IL-4, não produzem IgE e não apresentam hiper-responsividade brônquica, indicando o papel importante do STAT-6 nas respostas alérgicas.35
O GATA-3, um fator de transcrição seletivamente expresso pelos clones TH2, tem sido imputado na regulação da transcrição da IL-5. Estudos demonstraram que a expressão do GATA-3 mRNA em biópsias de pacientes com asma era maior quando comparada com biópsias de controles não asmáticos.36 O número de células que expressam transcrições GATA-3 tem correlação significante com o aumento da resistência e hiper-responsividade das vias aéreas em pacientes com asma. Estudos de colocalização evidenciaram que a maioria (aproximadamente 60 a 90%) de células GATA-3 mRNA+ nos brônquios de asmáticos era constituída de células T CD3(+), e em menor quantidade por eosinófilos MBP-positivos e mastócitos triptase-positivos. A densidade celular GATA-3 mRNA+ se correlacionava significativamente com o número de células que expressavam a citocina TH2 IL-5 mRNA.
Pela expressão de um mutante dominante negativo do GATA-3 em células T de modelo murino, se obtém a redução dos níveis de citocinas TH2 IL-4, IL-5 e IL-13.37 Em consequência, a eosinofilia brônquica, a produção de muco e a síntese de IgE sofrem considerável atenuação no rato transgênico. Baseados nestes estudos, pode-se demonstrar que o silenciamento do fator de transcrição comum GATA-3 é suficiente para abrandar respostas TH2 in vivo, oferecendo a opção de capturar todas citocinas de uma só vez (IL-4, IL-5 e IL-13) e assim, prevenir precocemente o início da cascata inflamatória.
Existem outros fatores de transcrição importantes na diferenciação TH1/TH2. O proto-oncogene c-maf, semelhante ao GATA-3, é também seletivamente expresso nos clones TH2, sendo induzido durante a diferenciação TH2 e não na TH1.38
Referências
01.Barnes PJ. Cytokines as mediators of chronic asthma. Am J Crit Care Med 1994; 150: S42-9.
02.Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann NY Acad Sci 1998; 851:139-46.
03.Adocck IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 2001; 14:211-9.
04.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307-13.
05.Struhl K, Moqtaderi Z. The TAFs in the HAT. Cell 1998; 94:1-4.
06.Pfitzner E, Jahne R, Wissler M, Stoecklin E, Groner B. p300/CREB-binding protein enhances the prolactin-mediated domain of Stat5, but does not participate in the Stat5-mediated supression of the glicocorticoid response. Mol Endocrinol 1998; 12:1582-93.
07.Zhu M, John S, Berg M, Leonard WJ. Functional association of Nmi with Stat - 1 in IL-2 and IFNgama-mediated signaling. Cell 1999; 96:121-30.
08.Barnes PJ, Adcock IM. Anti-inflammatory actions of steroids: molecular mechanisms. Trends Pharmacol Sci 1993; 14:436-41.
09.Blackwell TS, Christman JW. The role of nuclear factor kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol 1997; 17:3-9.
10.Christman JW, Lancaster LH, Blackwell TS.
Nuclear factor kappa B: a pivotal role in the systemic response syndrome and new target for therapy. Intensive Care Med 1998; 24:1131-8.
11.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kB. Ann Rev Cell Biol 1994; 10:405-55.
12.Lenardo MJ, Baltimore D. NF-kB: a pleiotropic mediator of inducible and tissue-specific gene control. Cell 1989; 58:227-9.
13.Barnes PJ, Adock IM. NF-kB: a pivotal role in asthma and a new target for therapy. Trends Pharmacol Sci 1997; 18:46-50.
14.Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kB. EMBO J 1991; 10:3805-17.
15.Stacey MA, Sun G, Vassalli G et al. The allergen Der p1 induced NF-kB activation through interference with IkB alpha function in asthmatic bronchial epithelial cells. Biochem Biophys Res Commun 1997; 236:522-6.
16.Liu SF, Haddad EB, Adcock I et al. Inducible nitric oxide synthase after sensitization and allergen challenge of Brown Norway rat lung. Br J Pharmacol 1997; 121:1241-6.
17.Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kB-mediated transcription. J Biol Chem 1999; 274:9707-20.
18.Yang L, Cohn L, Zhang DH, Homer R, Ray A, Ray P. Essential role of nuclear factor kappaB in induction of eosinophilia in allergic airway inflammation. J Exp Med 1998; 188:1739-50.
19.Fujisawa T, Chang MM, Velichko S, Thai P, Hung LY, Huang F, Phuong N, Chen Y, Wu R. NF-?B mediates IL-1ß- and IL-17A-induced MUC5B expression in airway epithelial cells. Am J Respir Cell Mol Biol 2011; 45:246-52.
20.Broide DH, Lawrence T, Doherty T, Cho JY, Miller M, McElwain K, McElwain S, Karin M. Allergen-induced peribronchial fibrosis and mucus production mediated by IkappaB kinase beta-dependent genes in airway epithelium. Proc Natl Acad Sci U S A 2005;102:17723-8.
21.Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL. Targeting the NF-kappaB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol Ther 2009; 121:1-13.
22.Janssen-Heininger YM, Poynter ME, Aesif SW, Pantano C, Ather JL, Reynaert NL, Ckless K, Anathy V, van der Velden J, Irvin CG, van der Vliet A. Nuclear factor kappaB, airway epithelium, and asthma: avenues for redox control. Proc Am Thorac Soc 2009; 6:249-55.
23.Menzel M, Akbarshahi H, Mahmutovic Persson I, Andersson C, Puthia M, Uller L. NF?B1 Dichotomously Regulates Pro-Inflammatory and Antiviral Responses in Asthma. J Innate Immun 2022; 14:182-191.
24.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kB and the glucocorticoid receptor. Proc Natl Acad Sci USA 1994; 91:752-6.
25.Adock IM, Shirasaki H, Gelder CM, et al. The effects of glucocorticoids on phorbol ester and cytokine stimulated transcription factor activation in human lung. Life Sci 1994; 55:1147-53.
26.Demoly P, Basset-Seguin N, Chanez P, Campbell AM, Gauthier-Rouvriere C, Godard P, Michel R, Bousquet J. C-fos proto-oncogene expression in bronchial biopses of asthmatics. Am J Resp Cell Mol Biol 1992; 7:128-33.
27.Adock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ. Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J Exp Med 1995; 182:1951-8.
28.Demoly P, Chanez P, Pujol JL, Gauthier-Rouvriere C, Michel FB, Godard P, Bousquet J. Fos immunoreactivity assessment on human normal and pathological bronchial biopsies. Respir Med 1995; 89:329-35.
29.Victoni T, Gleonnec F, Lanzetti M, Tenor H, Valença S, Porto LC, Lagente V, Boichot E. Roflumilast N-oxide prevents cytokine secretion induced by cigarette smoke combined with LPS through JAK/STAT and ERK1/2 inhibition in airway epithelial cells. PLoS One 2014; 9(1):e85243. doi: 10.1371/journal.pone.0085243.
30.O'Connell D, Bouazza B, Kokalari B, Amrani Y, Khatib A, Ganther JD, Tliba O. IFN-?-induced JAK/STAT, but not NF-?B, signaling pathway is insensitive to glucocorticoid in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2015; 309(4):L348-59.
31.Huang XP, Qin CY, Gao YM. miR-135a inhibits airway inflammatory response in asthmatic mice via regulating JAK/STAT signaling pathway. Braz J Med Biol Res 2021; 54(3):e10023. doi: 10.1590/1414-431X202010023.
32.Nikolskii AA, Shilovskiy IP, Barvinskaia ED, Korneev AV, Sundukova MS, Khaitov MR. Role of STAT3 Transcription Factor in Pathogenesis of Bronchial Asthma. Biochemistry (Mosc) 2021; 86:1489-1501.
33.Schindler C, Darnell JE. Transcriptional responses to polupeptide ligands: the JAK-STAT pathway. Annu Rev Biochem 1995; 64:621-51.
34.Mui ALF, Wakao H, O'Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO 1995; 14:1166-75.
35.Tomkinson A, Kanehiro A, Rabinovitch N, joetham A, Cieslewicz G, Gelfand EW. The failure of STAT6-deficient mice to develop airway eosinophilia and airway hyperresponsiveness is overcome by interleukin-5. Am J Respir Crit Care Med 1999; 160:1283-91.
36.Nakamura Y, Ghaffar O, Olivenstein R et al. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol 1999; 103:215-22.
37.Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray A. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity 1999; 11:473-82.
38.HO IC, Lo D, Glimcher LH. c-maf promotes T helper cell type 2 ( TH2) and attenuates TH1. ifferentiation by both interleukin-4 dependent and independent mechanisms. J Exp Med 1998; 188:1859-66.
|