|
Asma
GRANULOMATOSE EOSINOFÍLICA COM POLIANGIITE (GEPA) — (Síndrome de Churg-Strauss)
A GEPA é uma inflamação granulomatosa necrotizante rica em eosinófilos, muitas vezes acometendo o trato respiratório com vasculite necrotizante, afetando predominantemente vasos de pequeno porte, mas artérias e veias médias podem também ser comprometidas. Está associada à asma, eosinofilia e ao
anticorpo anticitoplasma de neutrófilos (ANCA).1
A GEPA é uma doença primariamente induzida por células Th2, resultando em alta produção de
citocinas inflamatórias características desse tipo. No entanto, a doença é bem mais complexa e inclui também outros tipos de células T, como as Tregs, Th17, o sistema inato e a desregulação humoral e de células B.
A GEPA era anteriormente conhecida como Síndrome de Churg-Strauss (SCS). Foi descrita pela primeira vez em 1951 pelo
Dr. Jacob Churg e pela Dra. Lotte Strauss
os quais relataram 13 casos que apresentavam a asma como primeira manifestação.2 Em 1990, o American College of Rheumatology (ACR) relaxou os critérios para a classificação, retirando a exigência da confirmação histopatológica de vasculite e granulomatose e publicou os critérios diagnósticos da síndrome, que devem incluir pelo menos quatro das seis de suas características, resultando em sensibilidade de 85% e especificidade de 99,7% (Tabela 1).3
Tabela 1 Critérios para o Diagnóstico da GEPA / SCS
• |
Asma moderada a severa |
• |
Anormalidade nos seios da face |
• |
Eosinofilia periférica >10% ou contagem absoluta >1.500 mm3 |
• |
Mononeuropatia ou polineuropatia |
• |
Infiltrados pulmonares |
• |
Presença extravascular de eosinófilos |
Em 1994 a síndrome foi definida como um distúrbio caracterizado por inflamação granulomatosa rica em eosinófilos no trato respiratório e vasculite necrosante de vasos de pequeno a médio calibre, associada à asma e à eosinofilia.4 Em 2012, a Nomenclatura Revisada de Vasculites da Conferência de Consenso de Chapel Hill recomendou o termo granulomatose eosinofílica com poliangiite (GEPA), acrescentando o conceito de associação da positividade do ANCA ao comprometimento renal na GEPA.5
Em 2022 o Diagnostic and Classification Criteria for Vasculitis (DCVAS), um estudo observacional multinacional com o objetivo de desenvolver critérios diagnósticos e atualizar critérios de classificação em vasculite definiu critérios ponderados aprovados pelo (ACR-EULAR) para a classificação da vasculite de pequenos e médios vasos, incluindo a GEPA.6
A patogênese da GEPA não é bem conhecida: HLA-DRB1*04 e *07, HLA-DRB4 e haplótipo IL10.2 do gene promotor da IL-10 são os determinantes genéticos mais estudados.7-9 A incidência na população geral oscila entre 2,4 a 10 casos por milhão/ano,10-13 enquanto que em asmáticos oscila em torno de 60 casos/milhão/ano.13 A taxa de sobrevida em cinco anos para a GEPA é de 60–97%.14
Mais de 90% dos pacientes com GEPA apresentam simultaneamente asma que, geralmente se inicia na idade adulta, é severa, dependente de corticoides, tendendo a piora progressiva, que antecede o início da doença sistêmica por vários anos (7–8 anos em média).15,16 Além da asma, os pacientes com GEPA apresentam caracteristicamente sintomas das vias aéreas superiores – rinossinusite crônica (47–93%) e pólipos nasais (62–77%)17 e sintomas gerais, como artralgia, mialgia, mal-estar, febre, mialgias e perda de peso.10,18 Mononeurite múltipla, geralmente sensorial, é descrita em 50–70% dos casos,19,20 neuropatia periférica, enquanto as lesões cutâneas são descritas em aproximadamente 60% dos pacientes através de rash macular ou papular, nódulos, lesões hemorrágicas. A púrpura palpável é um achado comum nas extremidades inferiores.21 O comprometimento renal ocorre em cerca de 40% dos pacientes através de glomerulonefrite e nefrite intersticial.22 Após os pulmões, o coração é o órgão mais acometido, contribuindo com 48% dos óbitos, principalmente por infarto agudo do miocárdio, pericardite aguda ou constritiva.
A apresentação da doença é muito variável, sendo na maioria das vezes trifásica:16
| 1 – |
A fase prodrômica alérgica/atópica de rinossinusite e asma, em estágios variados, geralmente de início na idade adulta, na segunda ou terceira décadas. |
| 2 – |
A fase eosinofílica com eosinofilia periférica e infiltrados de tecido inflamatório rico em eosinófilos. |
| 3 – |
A fase de grave vasculite necrotisante terminal que geralmente afeta múltiplos órgãos, incluindo pele, pulmões, coração, trato gastrintestinal e sistema nervoso. |
A duração das distintas fases da doença apresenta ampla variabilidade interindividual. Em determinados casos, observa-se progressão acelerada em poucas semanas, ao passo que outros pacientes permanecem na fase prodrômica por períodos prolongados por vezes anos antes do surgimento de manifestações clínicas compatíveis com eosinofilia sistêmica.
A doença cursa com hemossedimentação elevada, níveis de IgE geralmente aumentados, sendo os anticorpos anticitoplasma dos neutrófilos (ANCA) positivos em ~40% dos casos de GEPA, sendo que este teste negativo não afasta a possibilidade de diagnóstico da doença.
ANCAs, ou autoanticorpos específicos para neutrófilos desempenham um papel importante na patogênese da vasculite associada a ANCA. Eles promovem a migração e desgranulação de neutrófilos na parede vascular, resultando na liberação de enzimas,
radicais livres e outros metabólitos tóxicos, que causam dano endotelial e inflamação vascular.23-25 São diretamente responsáveis pela ativação dos neutrófilos, pela lesão endotelial e pela perpetuação da resposta inflamatória. Pacientes que são ANCA negativos tendem a ter mais complicações cardiopulmonares enquanto que os positivos têm as manifestações clássicas de vasculite.26 O ANCA é mais frequente quando de glomerulonefrite.5 O ANCA pode ser detectado por imunofluorescência indireta, que essencialmente mostra padrões citoplasmáticos e perinucleares (c-ANCA e p-ANCA, respectivamente), mas o teste de referência para vasculite associada a ANCA é o ensaio imunoenzimático para PR3-ANCA ou MPO-ANCA. P-ANCA e MPO-ANCA são os tipos predominantes de ANCA positivo observados em pacientes com GEPA.1
Embora os eosinófilos representem aproximadamente 1% dos leucócitos do sangue periférico, eles têm a propensão de deixar a corrente sanguínea e migrar para tecidos inflamados. Os eosinófilos e seus mediadores são efetores críticos para asma e para GEPA onde especialmente se encontram muito elevados (≥ 1.500 mm3). São a principal fonte da interleucina-5 (IL-5) e expressam intensamente o receptor IL-5α em sua superfície, sendo que a sinalização IL-5 promove a proliferação, maturação, ativação e recrutamento de eosinófilos, sendo a IL-5 capaz de modular as funções de uma grande variedade de células imunes.26-29
Recentemente foi descrita que a hipereosinofilia induzida por dupilumabe, um inibidor da IL-4 e IL-13 utilizado no tratamento da asma e outras doenças atópicas, pode precipitar a GEPA e a pneumonia eosinofílica.30-32 Ainda não estão, entretanto, bem compreendidos os mecanismos subjacentes e os possíveis fatores predisponentes.
Na GEPA, tanto as vias Th1 como Th2 são ativadas e os eosinófilos contribuem para a lesão dos órgãos,33 entretanto, a GEPA é considerada principalmente uma doença de resposta Th2.
O envolvimento da via Th1 é evidenciado pelo aumento da concentração sérica de interferon-gama (IFN-γ) nesses pacientes.34 Números elevados de células Th17 e frequência reduzida de células Treg foram encontrados em pacientes com GEPA.35 Uma comparação de imunofenótipos de linfócitos em pacientes com GEPA mostrou que, além do aumento da atividade dos linfócitos T, eles se correlacionaram com o aumento de plasmócitos e linfócitos T auxiliares foliculares (Tfh), indicando que a ativação das células B está envolvida no desenvolvimento da GEPA.36
O quadro histopatológico clássico consiste em vasculite necrotizante, com infiltrado eosinofílico proeminente com reação granulomatosa extravascular em torno de focos necróticos com histiócitos dispostos radialmente e células gigantes paliçadas perto de pequenas artérias ou arteríolas e vasculite eosinofílica. Observa-se ainda edema rico em fibrina, leucócitos, granulomas sarcoides, fibrose focal e microabscessos eosinofílicos (Figura 1).37 Em outros órgãos pode ocorrer necrose fibrinoide de pequenos vasos que é a responsável pelos achados de glomerulonefrite, miosite e mononeurites.
Figura 1 Histopatologia da GEPA

Como a GEPA coexiste em pacientes com asma de difícil tratamento muitas vezes há dificuldade em se distinguir da asma grave 'verdadeira', daí a necessidade de investigações apropriadas no curso do tratamento da asma grave. Os exames de imagem contribuem para isso.
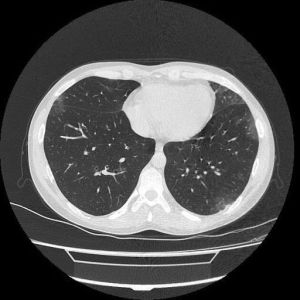
Os achados mais comuns na Tomografia Computadorizada do Tórax (TC) em pacientes com GEPA consistem em áreas de consolidação ou atenuação em vidro fosco, heterogêneas, de distribuição lobular, bilaterais, periféricas, migratórias, associadas com doença das vias aéreas (Figura 2).38 Pode haver espessamento septal interlobular, reticular intersticial ou reticulonodular.39
 |
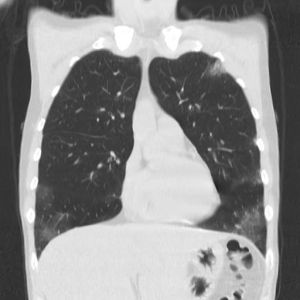 |
Figura 2 Jovem de 30 anos com GEPA, com tosse não produtiva crônica, febre baixa, relato de asma há dez anos. Tomografia computadorizada de alta resolução (TCAR) demonstrando infiltrados heterogêneos, bilaterais, em vidro fosco na periferia de ambos os pulmões. Cortesia de
Shervin Sharifkashani. From the case https://radiopaedia.org/cases/98647?lang=us">rID: 98647
|
A TC de tórax é útil para diferenciar a GEPA de asma grave. Um estudo revelou que a pontuação radiológica dos pacientes com GEPA foi significativamente maior em comparação aos pacientes com asma grave. Além disso, opacidades difusas em vidro fosco foram observadas em 74% dos pacientes com GEPA, enquanto apenas 18% dos pacientes com asma grave apresentaram essa característica (p<0,001).40,41
Tabela 2 Diagnóstico Diferencial da GEPA
DIAGNÓSTICO DIFERENCIAL DA GEPA
|
Doenças |
Exemplos Principais |
Semelhança com a GEPA |
Pontos de Distinção |
●Vasculites ANCA-associadas |
Granulomatose c/poliangiite (GPA) Poliangiite microscópica (PAM) |
Vasculite de pequenos vasos, ANCA (+) glomerulonefrite, envolvimento respiratório |
GEPA: eosinofilia marcante infiltrados pulmonares migratórios; GPA granulomas necrotizantes de vias aéreas; PAM ausência de eosinofilia/asma |
●Outras Vasculites Sistêmicas |
Poliarterite nodosa (PAN)
Arterite de Células Gigantes |
Comprometimento multissistêmico, febre, mialgia, neuropatia periférica |
GEPA: eosinofilia e atopia/asma; PAN acomete médios vasos sem eosinofilia/asma; ACG em idosos, cefaleia, claudicação da mandíbula |
●Pneumopatias Eosinofílicas |
Pneumonia Eosinofílica Crônica,42 Síndrome de Loeffler, Síndrome Hipereosinofílica43 |
Infiltrados pulmonares, eosinofilia periférica |
GEPA: vasculite sistêmica + ANCA (em parte dos casos); Pneumonia eosinofílica restrita ao pulmão; SH: ausência de vasculite |
●Doenças Infecciosas |
Parasitárias – Strongyloides, Áscaris; Micoses – Aspergillus - ABPA |
Eosinofilia, manifestações pulmonares, asma (no caso da ABPA) |
GEPA: vasculite sistêmica; infecções: sorologias positivas, história epidemiológica |
●Colagenoses |
Lúpus Eritematoso Sistêmico; Artrite Reumatoide c/Vasculite |
Vasculite, manifestações sistêmicas (cutâneas, articulares, rim) |
GEPA: eosinofilia, + asma; autoanticorpos, específicos (FAN, anti-DNA, FR, anti-ccp) |
●Doenças Alérgicas/Respiratórias |
Asma Grave Isolada / Rinite Alérgica Persistente |
Asma, rinite, eosinofilia |
GEPA: além da asma, presença da vasculite sistêmica e neuropatia periférica |
●Doenças Hematológicas/Neoplásicas |
Síndrome Hipereosinofílica Neoplásica44 – Leucemia Eosinofílica Crônica |
Eosinofilia persistente, envolvimento multissistêmico |
GEPA: antecedentes de atopia/asma, vasculite; Hemopatias achados medulares clonais |
Tratamento
Inicialmente deve ser salientado que, todo paciente com GEPA que apresenta asma, requer tratamento multidisciplinar na fase de diagnóstico assim como nas fases de acompanhamento clínico e terapêutico em conjunto com pneumologista, reumatologista e otorrinolaringologista, sendo que o tratamento com corticoide inalatório em combinação com β2-agonistas de longa ação deve sempre ser reavaliado e otimizado.
De acordo com as diretrizes baseadas em evidências para o diagnóstico e tratamento da GEPA publicado em 2023
o tratamento de indução de remissão deve ser adaptado às manifestações clínicas com relevância prognóstica.1 As manifestações de risco de órgãos, como insuficiência renal, proteinúria, cardiomiopatia, envolvimento gastrintestinal e do sistema nervoso central, além de neuropatia periférica e outras manifestações raras (como hemorragia alveolar), devem ser consideradas na escolha das estratégias de indução de remissão. É o chamado Escore de Cinco Fatores, que prediz o prognóstico e gravidade do quadro.45 O Escore de Cinco Fatores desenvolvido pelo grupo de vasculites Francês avalia a gravidade e prevê o prognóstico. Os seguintes fatores significativamente associados à mortalidade maior em 5 anos descritos são: 1) idade >65 anos; 2) insuficiência renal (creatinina >1,7 mg/dl); 3) insuficiência cardíaca; 4) envolvimento gastrintestinal (pancreatite, hemorragias, perfuração intestinal); 5) ausência de manifestações otorrinolaringológicas.
Em 2018 o grupo do Francês de Estudos de Vasculite (FVSG) descreveu a evolução da mortalidade ao longo do tempo pelas vasculites necrotizantes sistêmicas. Entre os 2.217 incluídos haviam 22,6% com GEPA.
A taxa de sobrevida em 5 anos aumentou de 72,2% (intervalo de confiança de 95% [IC] 59,7-87,2) para pacientes diagnosticados antes de 1980 para 94,5% (95% [IC] 90,4-98,8) após 2010 (p<0,001).
A taxa de mortalidade geral foi 2,26 por 100 pessoas-anos. Essa publicação pode demonstrar que a GEPA dentro do espectro das vasculites sistêmicas foi aquela que apresentou um melhor prognóstico em cinco anos, provavelmente devido aos pacientes poderem ser tratados antes que ocorresse dano irreversível.46
Para indução de remissão o corticoide é administrado como droga única. Entretanto, nos casos mais severos desde a década de 1970, a combinação de corticoides em altas doses e a ciclofosfamida ainda se mantém como o principal esquema terapêutico para o tratamento de casos graves,47 com a alternativa do rituximabe, cuja depleção de células B mostrou resultados favoráveis para indução de remissão.
Para pacientes com doença grave inicia-se com 500-1000 mg de metilprednisolona intravenoso por dia, por três dias, devendo prosseguir em curso oral de prednisolona 0,75–1 mg/kg/dia. A ciclofosfamida é introduzida em pulsos (600 mg/m2 pulso) inicialmente a cada duas semanas no primeiro mês, passando a mensal. Porém, tanto o número de doses quanto a sua duração ainda não estão plenamente estabelecidos, sendo o objetivo a indução da remissão completa. (Figura 3) Os esquemas pulsados são preferidos devido à menor dose total de ciclofosfamida administrada e ao menor risco de complicações relacionadas à bexiga e menor taxa de leucopenia.48
O mepolizumabe apresenta indicação na GEPA na fase de manutenção da remissão, sobretudo em pacientes com asma grave,
facilitando a redução na dose de corticoides. O mepolizumabe é um anticorpo monoclonal IgG1k murino humanizado. O mepolizumabe se liga à IL-5
prevenindo sua associação com a cadeia alfa do complexo receptor de IL-5 na superfície da célula eosinofílica. Como consequência, a diferenciação, ativação e crescimento de eosinófilos são inativados. Wechsler et al.49 em
estudo multicêntrico, duplo-cego, de grupos paralelos avaliou pacientes
participantes com granulomatose eosinofílica recidivante ou refratária. O estudo demonstrou benefícios do mepolizumabe em relação aos dois desfechos primários: as semanas acumuladas de remissão da doença e a proporção de participantes que estavam em remissão na semana 36 e na semana 48 do estudo
(32% vs. 3%), além da redução no uso do corticoide. A dose recomendada é de 300 mg de mepolizumabe administrada por injeção subcutânea (SC) uma vez a cada 4 semanas.
Embora o tratamento prolongado com corticoide oral (CO) reduza o risco de recidiva, ele está associado a efeitos de dose cumulativa progressiva e à toxicidade intrínseca propriamente dita.50,51
Um estudo de longo prazo do mepolizumabe para avaliação de efetividade e segurança no mundo real de GEPA no Japão (MARS) evidenciou, para um período de 144 semanas, redução da dose de CO em paralelo ao controle da doença em comparação ao pré-tratamento entre pacientes com GEPA. Nas semanas 45–48, mais de um terço dos pacientes cerca de 38% não mais necessitavam de tratamento com CO (62% receberam CO) e a dose média de CO foi de 3,3 mg/dia (considerada muito baixa).52 Reduções relevantes de ≥ 50% na dose de COs também foram observadas em cerca de 57% dos pacientes em mepolizumabe versus 21% em placebo, em uma análise post hoc do estudo MIRRA.53
Em estudo mais recente Wechsler et al.54 efetuaram ensaio clínico multicêntrico, duplo-cego, randomizado, controlado por ativo, de não inferioridade, para avaliar a eficácia e a segurança do benralizumabe em comparação ao mepolizumabe. Avaliaram 140 pacientes com GEPA recidivante e refratária em dois grupos na proporção 1:1 (70 atribuídos a cada fármaco). O percentual ajustado com remissão nas semanas 36 e 48 foi de 58% no grupo benralizumabe e 59% no mepolizumabe, denotando não inferioridade, mas não superioridade do benralizumabe em relação ao mepolizumabe. Quanto à suspensão dos COs durante as semanas 48 a 52, parcela de 41% alcançou retirada completa utilizando benralizumabe, contra 26% daqueles que receberam mepolizumabe. O benralizumabe não foi inferior ao mepolizumabe para a indução de remissão em pacientes com doença recidivante ou refratária, mostrando não inferioridade, mas não mostrando superioridade sob o ponto de vista estatístico. Eventos adversos ocorreram em 90% dos pacientes no grupo benralizumabe e 96% do mepolizumabe, entretanto eventos graves foram relatados em 6% e 13% respectivamente.
Cabe definir remissão da GEPA como sendo
caracterizada pela ausência de sinais ou sintomas clínicos atribuíveis à doença ativa, incluindo asma e manifestações otorrinolaringológicas. Além disso, a dose diária de corticoides deve ser considerada para definir a remissão com dose máxima de 7,5 mg de prednisolona/dia como sendo um possível ponto de corte. Também é recomendável definir remissão baseada no escore Birmingham Vasculitis Activity Score (BVAS) de zero. No caso de tratamento concomitante de corticoide da mesma forma se aceita uma dose máxima equivalente de prednisolona de 7,5 mg por dia.
Referências
01.Emmi G, Bettiol A, Gelain E, Bajema IM, Berti A, Burns S, Cid MC, Cohen Tervaert JW, Cottin V, Durante E, Holle JU, Mahr AD, Del Pero MM, Marvisi C, Mills J, Moiseev S, Moosig F, Mukhtyar C, Neumann T, Olivotto I, Salvarani C, Seeliger B, Sinico RA, Taillé C, Terrier B, Venhoff N, Bertsias G, Guillevin L, Jayne DRW, Vaglio A. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol 2023; 19:378-393.
02.Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 1951; 27:277-301.
03.Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33:1094-100.
04.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CGM, McCluskey RT, Sinico RA, Rees AJ, van Es LA, Waldherr R, Wiik A. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum. 1994; 37 :187-192.
05.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013; 65:1-11.
06.Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, Khalid S, Hutchings A, Luqmani RA, Watts RA, Merkel PA; DCVAS Study Group. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann Rheum Dis 2022; 81:309-314.
07.Gioffredi A, Maritati F, Oliva E, Buzio C. Eosinophilic granulomatosis with polyangiitis: an overview. Front Immunol 2014; 5:549. doi:10.3389/fimmu.2014.00549.
08.Wieczorek S, Hellmich B, Arning L, Moosig F, Lamprecht P, Gross WL, Epplen JT. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener's granulomatosis. Arthritis Rheum 2008; 58:1839-48.
09.Vaglio A, Martorana D, Maggiore U, Grasselli C, Zanetti A, Pesci A, Garini G, Manganelli P, Bottero P, Tumiati B, Sinico RA, Savi M, Buzio C, Neri TM; Secondary and Primary Vasculitis Study Group. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum 2007; 56:3159-66.
10.De Caterina R, Zampolli A. From asthma to atherosclerosis 5-Lipoxygenase, leukotrienes, and inflammation. N Engl J Med 2004; 350:4.
11.Malmstrom K, Rodriguez-Gomez G, Guerra J, et al. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma. A randomized controlled trial. Ann Intern Med 1999; 130:487-495.
12.Margaritopoulos GA, Wells AU. – Pulmonary Vasculitis. In: Palange P, Rohde G. Respiratory Medicine. Sheffield: Latimer; 2019: 637-645.
13.Silverman ES, Du J, De Sanctis GT, Rådmark O, Samuelsson B, Drazen JM, Collins T. Egr-1 and Sp1 interact functionally with the 5-lipoxygenase promoter and its naturally occurring mutants. Am J Respir Cell Mol Biol 1998; 19:316-323.
14.
Mukhtyar C, Flossmann O, Hellmich B, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67:1004-10.
15.Cottin V, Bel E, Bottero P, et al. Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur Respir J 2016; 48:142941.
16.Frankel SK, Schwarz MI. The pulmonary vasculitides. Am J Respir Crit Care Med. 2012; 186:216-24.
17.Chakraborty RK, Rout P. Eosinophilic Granulomatosis With Polyangiitis (Churg-Strauss Syndrome). 2024 Sep 19. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan. PMID: 30725784.
18.Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, Ruoppolo G, Altissimi G, De Vincentiis M. Churg-Strauss syndrome. Autoimmun Rev 2015; 14:341-8.
19.Cho HJ, Yune S, Seok JM, Cho EB, Min JH, Seo YL, Lee BJ, Kim BJ, Choi DC. Clinical Characteristics and Treatment Response of Peripheral Neuropathy in the Presence of Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss Syndrome): Experience at a Single Tertiary Center. J Clin Neurol 2017; 13:77-83.
20.Padoan R, Marconato M, Felicetti M, Cinetto F, Cerchiaro M, Rizzo F, Marcolongo R, Punzi L, Agostini C, Schiavon F. Overall Disability Sum Score for Clinical Assessment of Neurological Involvement in Eosinophilic Granulomatosis With Polyangiitis. J Clin Rheumatol 2018; 24:197-202.
21.Davis MD, Daoud MS, McEvoy MT, Su WP. Cutaneous manifestations of Churg-Strauss syndrome: a clinicopathologic correlation. J Am Acad Dermatol 1997; 37:199-203.
22.Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, Maurier F, Jouneau S, Bienvenu B, Puéchal X, Aumaître O, Le Guenno G, Le Quellec A, Cevallos R, Fain O, Godeau B, Seror R, Dunogué B, Mahr A, Guilpain P, Cohen P, Aouba A, Mouthon L, Guillevin L; French Vasculitis Study Group. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 2013; 65:270-81.
23.Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med 1997; 337:1512-23.
24.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 1988; 318:1651-7.
25.Hilhorst M, van Paassen P, Tervaert JW; Limburg Renal Registry. Proteinase 3-ANCA Vasculitis versus Myeloperoxidase-ANCA Vasculitis. J Am Soc Nephrol 2015; 26:2314-27.
26.Kallenberg CG. Pathogenesis of ANCA-associated vasculitides. Ann Rheum Dis 2011 ;70: i59-i63.
27.Pavord ID, Bel EH, Bourdin A, Chan R, Han JK, Keene ON, Liu MC, Martin N, Papi A, Roufosse F, Steinfeld J, Wechsler ME, Yancey SW. From DREAM to REALITI-A and beyond: Mepolizumab for the treatment of eosinophil-driven diseases. Allergy 2022; 77:778-797.
28.Pelaia C, Paoletti G, Puggioni F, Racca F, Pelaia G, Canonica GW, Heffler E. Interleukin-5 in the Pathophysiology of Severe Asthma. Front Physiol 2019; 10:1514.
29.Varricchi G, Bagnasco D, Borriello F, Heffler E, Canonica GW. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr Opin Allergy Clin Immunol 2016; 16:186-200.
30.Yamazaki K, Nomizo T, Hatanaka K, Hayama N, Oguma T, Asano K. Eosinophilic granulomatosis with polyangiitis after treatment with dupilumab. J Allergy Clin Immunol Glob 2022; 1:180-182.
31.Suzaki I, Tanaka A, Yanai R, Maruyama Y, Kamimura S, Hirano K, Kobayashi H. Eosinophilic granulomatosis with polyangiitis developed after dupilumab administration in patients with eosinophilic chronic rhinosinusitis and asthma: a case report. BMC Pulm Med 2023; 23(1):130.
32.Nishiyama Y, Koya T, Nagano K, Abe S, Kimura Y, Shima K, Toyama-Kosaka M, Hasegawa T, Sasaki T, Shinbori K, Ueki S, Takamura K, Kikuchi T. Two cases of dupilumab-associated eosinophilic pneumonia in asthma with eosinophilic chronic rhinosinusitis: IL-5-driven pathology? Allergol Int 2022; 71:548-551.
33.Trivioli G, Terrier B, Vaglio A. Eosinophilic granulomatosis with polyangiitis: understanding the disease and its management. Rheumatology (Oxford). 2020; 59(Suppl 3):iii84-iii94.
34.Khoury P, Grayson P, Klion A. Eosinophils in vasculitis: characteristics and roles in pathogenesis. Nat Rev Rheumatol 2014; 10:474-83.
35.Fijolek J, Radzikowska E. Eosinophilic granulomatosis with polyangiitis - Advances in pathogenesis, diagnosis, and treatment. Front Med 2023; 10:1145257.
36.Kubo S, Kanda R, Nawata A, Miyazaki Y, Kawabe A, Hanami K, Nakatsuka K, Saito K, Nakayamada S, Tanaka Y. Eosinophilic granulomatosis with polyangiitis exhibits T cell activation and IgG4 immune response in the tissue; comparison with IgG4-related disease. RMD Open 2022; 8(1):e002086.
37.Weisenberg. Elliot. Pathology Outlines – Eosinophilic granulomatous with poluangiitis (EGPA). 2024. Disponível em: www.pathologyoutlines.com/topic/lungnontumorallergicgran.html
38.Worthy SA, Muller NL, Hansell DM, Flower CD. Churg-Strauss syndrome: the spectrum of pulmonary CT findings in 17 patients. Am J Roentgenol 1998; 170:297-300.
39.Lin X, Lin Y, Lai Z, Wei S, Qiu M, Li J, Liu Q, Chung KF, Zeng Q, Zhang Q. Retrospective comparison of high-resolution computed tomography of eosinophilic granulomatosis with polyangiitis with severe asthma. Ann Transl Med 2021; 9:983. doi:10.21037/atm-21-2243.
40.Lin X, Lin Y, Lai Z, Wei S, Qiu M, Li J, Liu Q, Chung KF, Zeng Q, Zhang Q. Retrospective comparison of high-resolution computed tomography of eosinophilic granulomatosis with polyangiitis with severe asthma. Ann Transl Med 2021; 9:983. doi:10.21037/atm-21-2243.
41.Aigbirior J, Almaghrabi A, Lafi M, Mansur AH. The role of radiological imaging in the management of severe and difficult-to-treat asthma. Breathe 2024; 20: 240033; DOI: 10.1183/20734735.0033-2024.
42.
Marchand E, Etienne-Mastroianni B, Chanez P, Lauque D, Leclerc P, Cordier JF; Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires. Idiopathic chronic eosinophilic pneumonia and asthma: how do they influence each other? Eur Respir J 2003l; 22:8-13.
43.Requena G, van den Bosch J, Akuthota P, Kovalszki A, Steinfeld J, Kwon N, Van Dyke MK. Clinical Profile and Treatment in Hypereosinophilic Syndrome Variants: A Pragmatic Review. J Allergy Clin Immunol Pract 2022; 10:2125-2134.
44.Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, Hellmann A, Metzgeroth G, Leiferman KM, Arock M, Butterfield JH, Sperr WR, Sotlar K, Vandenberghe P, Haferlach T, Simon HU, Reiter A, Gleich GJ. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130:607-612.e9.
45.Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Toumelin PL; French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011 Jan;90(1):19-27.
46.Jardel S, Puéchal X, Le Quellec A, Pagnoux C, Hamidou M, Maurier F, Aumaitre O, Aouba A, Quemeneur T, Subra JF, Cottin V, Sibilia J, Godmer P, Cacoub P, Fauchais AL, Hachulla E, Maucort-Boulch D, Guillevin L, Lega JC; French Vasculitis Study Group (FVSG). Mortality in systemic necrotizing vasculitides: A retrospective analysis of the French Vasculitis Study Group registry. Autoimmun Rev 2018; 17:653-659.
47.Novack SN, Pearson CM. Cyclophosphamide therapy in Wegener's Granulomatosis. N Engl J Med 1971; 284:938-42.
48.de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, Luqmani R, Pusey CD, Rasmussen N, Sinico RA, Tesar V, Vanhille P, Westman K, Savage CO; EUVAS (European Vasculitis Study Group). Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009; 150:670-80.
49.Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, Merkel PA, Moosig F, Specks U, Cid MC, Luqmani R, Brown J, Mallett S, Philipson R, Yancey SW, Steinfeld J, Weller PF, Gleich GJ; EGPA Mepolizumab Study Team. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med 2017; 376:1921-1932.
50.Daugherty J, Lin X, Baxter R, Suruki R, Bradford E. The impact of long-term systemic glucocorticoid use in severe asthma: a UK retrospective cohort analysis. J Asthma 2018;55:651-658.
51.Strehl C, Bijlsma JW, de Wit M, Boers M, Caeyers N, Cutolo M, Dasgupta B, Dixon WG, Geenen R, Huizinga TW, Kent A, de Thurah AL, Listing J, Mariette X, Ray DW, Scherer HU, Seror R, Spies CM, Tarp S, Wiek D, Winthrop KL, Buttgereit F. Defining conditions where long-term glucocorticoid treatment has an acceptably low level of harm to facilitate implementation of existing recommendations: viewpoints from an EULAR task force. Ann Rheum Dis 2016; 75:952-7.
52.Price DB, Trudo F, Voorham J, Xu X, Kerkhof M, Ling Zhi Jie J, Tran TN. Adverse outcomes from initiation of systemic corticosteroids for asthma: long-term observational study. J Asthma Allergy 2018; 11:193-204.
53.Steinfeld J, Bradford ES, Brown J, Mallett S, Yancey SW, Akuthota P, Cid MC, Gleich GJ, Jayne D, Khoury P, Langford CA, Merkel PA, Moosig F, Specks U, Weller PF, Wechsler ME. Evaluation of clinical benefit from treatment with mepolizumab for patients with eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol 2019; 143:2170-2177.
54.Wechsler ME, Nair P, Terrier B, Walz B, Bourdin A, Jayne DRW, Jackson DJ, Roufosse F, Börjesson Sjö L, Fan Y, Jison M, McCrae C, Necander S, Shavit A, Walton C, Merkel PA; MANDARA Study Group. Benralizumab versus Mepolizumab for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med 2024; 390:911-921.
|