|
Asma
Tratamento da Asma
ASMA ESTEROIDE RESISTENTE
Embora mais de 90% dos pacientes respondam ao uso regular de corticoides inalados, uma pequena proporção constituída por doentes com asma crônica necessita de suplementação oral diária ou em dias alternados, sendo rotulados como Asmáticos Esteroide-Dependentes (AED). Antes de qualificá-los como AED, deve-se avaliar a adesão do paciente ao tratamento e descartar anormalidades farmacocinéticas, tais como a rápida eliminação da droga, a possibilidade de a dose livrada não alcançar o sítio de ação, a incompleta absorção do corticoide por via oral ou a má técnica de utilização do dispositivo utilizado para livrar a droga.
Existem pacientes que não conseguem controlar sua asma nem mesmo com altas doses de prednisolona oral, sendo rotulados como Asmáticos esteroide resistentes (AER). Antes de serem qualificados como AER, deve-se afastar as causas de asma secundária: a aspirina, a vasculite, a aspergilose alérgica broncopulmonar e a decorrente de obstrução brônquica irreversível. Muitas vezes uma obstrução de vias aéreas superiores, secundária à disfunção de cordas vocais, pode mimetizar a asma. Esta patologia pode ser descartada através da execução da curva fluxo-volume, ou por endoscopia, quando se visualiza a adução paradoxal das cordas vocais na inspiração.
A verdadeira AER é rara, ocorrendo em apenas 1:1.000 ou 1:10.000 pacientes com asma.1,2 Estes são definidos pela incapacidade que apresentam de melhorar seu VEF1 matinal, pré-broncodilatador (< 70–80% do teórico), em pelo menos 15% do basal prévio, após um ciclo de 7 a 14 dias de uso contínuo de 30–40 mg/dia em tomada única matinal de prednisolona oral ou outra droga em dose equivalente. Este teste deve ser precedido por um período de duas semanas de utilização de placebo.3 Estes pacientes, entretanto, quando testados com broncodilatadores de ação rápida, apresentam melhora no VEF1 superior a 15%, o que os distingue da obstrução fixa da doença obstrutiva crônica ou do remodelamento das vias aéreas da asma severa. A biópsia brônquica por fibroscopia em pacientes com AER evidencia um infiltrado inflamatório eosinofílico semelhante ao encontrado em asmáticos sensíveis ao corticoide.4 Deve ser ressaltado que asmáticos de difícil controle não são necessariamente esteroide resistentes.
A resistência ao corticoide, também chamada de síndrome de Chrousos, é encontrada em outras doenças inflamatórias e em doenças de causa imunológica como nas doenças intestinais inflamatórias, no lúpus eritematoso sistêmico e na artrite reumatoide. Torna-se importante a distinção entre a resistência ao corticoide das doenças inflamatórias e a rara síndrome endócrina conhecida como Resistência Familiar ao Corticoide (RFC). A RFC caracteriza-se por altos níveis circulantes de cortisol e do Hormônio Adrenocorticotrófico (ACTH), com ausência de sinais e sintomas da síndrome de Cushing e sinais e sintomas de excesso de androgênio (hirsutismo e alterações menstruais nas mulheres).
Ao contrário da síndrome de Cushing, em pacientes com síndrome de Chrousos, o eixo hipotálamo-hipófise-adrenal preserva seu ritmo circadiano.5 A RFC ocorre por provável anormalidade estrutural do Receptor de Glicocorticoide (GR),6 tendo sido descritas várias alterações na função dos GRs nos leucócitos do sangue periférico e também em fibroblastos destes pacientes. Estas alterações incluem a redução na afinidade do GR pelo cortisol, a redução no número dos GRs, a termolabilidade do GR, e anormalidades na ligação do complexo GR com o DNA.
Até o momento, foram identificadas mais de 15 diferentes mutações do GR que causam resistência aos corticoides.7
A resistência aos efeitos anti-inflamatórios e imunomoduladores do corticoide difere da RFC por não estar associada a elevadas concentrações de cortisol ou ACTH, não ocorrendo hipertensão arterial, hipopotassemia ou elevação de androgênios.
Quanto aos mecanismos da resistência, podem estar relacionados à baixa afinidade ou redução nos sítios de ligação dos GRs por célula, além de outras anormalidades na resposta imunocelular relacionada aos linfócitos T (com persistente ativação não responsiva ao corticoide), eosinófilos e monócitos.
Existem dois tipos de asma esteroide resistente: a forma adquirida ou Tipo I e a forma primária ou Tipo II.8,9 A maioria dos pacientes com AER se inclui na do Tipo I. Estes pacientes desenvolvem os efeitos colaterais inerentes ao uso crônico e em altas doses da droga, como a osteoporose, a supressão das adrenais e a síndrome de Cushing iatrogênica. Isto ocorre pois nos pacientes com AER do Tipo I a resistência aos corticoides acontece somente nas células imunoinflamatórias (p. ex. células T). O resto dos tecidos orgânicos permanece sensível aos efeitos deletérios da terapêutica sistêmica da droga.
Os corticoides atuam reprimindo os genes inflamatórios, por inibição direta dos fatores de transcrição AP-1 (activator protein-1) e NF-kB (fator nuclear-kB), que aumentam a expressão de genes inflamatórios (citocinas, adesão de moléculas, quimiotáticos, enzimas e receptores).
O mecanismo molecular da AER foi demonstrado por uma reduzida interação (afinidade) entre o corticoide e os ligantes dos GRs das células mononucleares, em especial os linfócitos T, ou as sequências DNA específicas dos genes corticoide responsivos, conhecidos também como elementos de resposta dos corticoides (GREs).10,11 Estas deficiências de afinidade podem ser devidas ao aumento da expressão da isoforma GR-ß que interfere com a ligação da isoforma ativa GR-a aos GREs, no núcleo celular. Os GR-ß estão localizados no núcleo das células, independente do tratamento com corticoides. Ao contrário do GR-a, o GR-ß é incapaz de ligar-se aos corticoides e incapaz de ativar genes corticoide sensíveis. O GR-ß apresenta um domínio DBD intacto e é capaz de se ligar ao DNA ao nível dos GREs. O GR-ß pode inibir a atividade GR-a através da ligação com o GR-a como um heterodímero ao nível dos GREs no promotor dos genes GR-regulados. Acredita-se que esta insensibilidade aos corticoides em pacientes com AER possa ser mediada pelos níveis persistentemente elevados de citocinas IL-2 e IL-4 encontradas nas vias aéreas destes pacientes. Estas alterações de ligação dos GRs podem ser revertidas quando estas células são incubadas em meio de cultura sem citocinas.9,12 Ainda é desconhecido o exato mecanismo pelo qual as citocinas, como a combinação da IL-2 e IL-4, reduzem a responsividade dos corticoides em paciente com AER tipo I.
Na AER do Tipo II a resistência aos corticoides é generalizada, afetando todos os tecidos, estando associada a uma mutação no gene do GR ou nos genes que modulam a sua função. Ocorre um defeito irreversível do GR envolvendo todos os tipos de célula.
Na AER do Tipo II, os pacientes não apresentam deficiência na afinidade com os GRs, porém o número destes é extremamente baixo, o que parece ser uma alteração irreversível. Estes pacientes são refratários aos efeitos colaterais dos corticoides, sugerindo que o baixo número de GRs ocorra também em outros tecidos.
Outras possíveis causas para a AER incluem:13,14 ativação de citocina pró-inflamatória do p38 mitogen-activated protein kinase que interfere com a translocação nuclear do receptor de corticoide; a formação de autoanticorpos contra a lipocortin-1 (algumas das ações anti-inflamatórias dos corticoides podem ser devidas à indução da lipocortin); falha na acetilação do resíduo lisina da histona-4 da cromatina, com redução na ativação de genes anti-inflamatórios e aumento na expressão do receptor de corticoide GR-ß que funciona como inibidor dominante negativo, competindo com o GR-a; citocinas TH2 têm participação em parte na refratariedade dos corticoides na asma, quando os linfócitos CD4+ tornam-se incapazes de produzir a citocina anti-inflamatória IL-10, em resposta à dexametasona, o que não ocorre em pacientes sensíveis aos corticoides;15 participação genética na redução da sensibilidade dos corticoides pôde ser avaliada por Weiss et al.16 que examinaram 31 polimorfismos de base única (SNP) em 14 genes candidatos em pacientes com asma e identificaram um gene, o CRHR-1 (corticotrophin-releasing hormone receptor-1) que continha polimorfismo associado a menos responsividade em três diferentes populações; downregulation do receptor de corticoide; altas doses de ß2-agonistas, que teoricamente podem reduzir a responsividade do corticoide na asma, e afetar pacientes suscetíveis; infecções virais, pois os vírus podem ativar fatores de transcrição, resultando em resistência ao corticoide.
Segundo Nicola Hanania17 os possíveis mecanismos celulares associados à resistência aos corticoides na asma incluem:18-20
Conceitos Atuais sobre Possíveis Mecanismos Celulares Associados |
à Resistência aos Corticoides na Asma |
| |
Disfunção ou anomalias genéticas do receptor citoplasmático de glicocorticoide-a (GR-a) podem ocorrer devido a: a) aumento da fosforilação de cinases (como a proteína cinase ativada por mitógeno p38 a e/ou γ e a cinase I c-Jun-N-terminal), resultando em diminuição da translocação nuclear; b) elevação de IL-2, IL-4 e IL-13, que reduz a fosforilação do GR-a e, consequentemente, a translocação nuclear; e c) aumento da sintase do óxido nítrico induzível. |
| |
Aumento de GR-ß em células epiteliais, em decorrência do aumento de IL-17. |
| |
Acetilação da histona face à redução da histona desacetilase 2, com diminuição da repressão do gene inflamatório. |
| |
Aumento da expressão de fatores de transcrição pró-inflamatórios: AP-1 e NF-kB. |
Estudos em animais identificaram novos mecanismos de resistência aos corticoides, incluindo: expressão alterada dos microRNAs 9 e 21;
maior expressão de inflamassomas de proteína-3 do receptor nod-like/IL-1ß, TNF-a, interferon γ (IFN-γ), ligante indutor de apoptose relacionado ao TNF, TSLP;
aumento da sinalização de fosfatidilinositol-3-quinase (PI3K) e receptores Toll-like (TLR);
aumento das espécies reativas de oxigênio (ROS);
exposição a alérgenos como ácaros e ovoalbumina;
infecções por patógenos como clamídia e vírus, como HRV-IB, RSV e Aspergillus;
o fator de crescimento transformador-ß (TGF-ß) desregulando o equilíbrio entre GR-a e GR-ß; e IL-33. Além do mais, poluição do ar, dieta rica em gordura e obesidade também foram.19,21
Diagnóstico da Asma Esteroide Resistente
Cerca de 5–10% dos pacientes com asma enquadram-se no grupo de asma grave. Destes, cerca de 75% são sensíveis ao corticoide, enquanto que 25% são resistentes. Geralmente são pacientes com evolução clínica desfavorável, em que ocorrem sérios efeitos adversos da droga, em decorrência das altas doses impostas, sem que haja resposta terapêutica adequada. O manejo destes pacientes impõe uma abordagem detalhada, objetivando afastar outras patologias e possíveis causas de falência terapêutica. Citamos:
Anamnese detalhada, exame físico e provas de função respiratória para confirmar o diagnóstico de asma. Considerar outras patologias como: obstrução laríngea induzível, antes conhecida como disfunção das cordas vocais, traqueomalácia, refluxo gastresofagiano e sinusite crônica.
Identificar alérgenos que possam desencadear ou perenizar a doença, instituindo-se o controle ambiental apropriado. Pacientes com asma crônica, alérgicos aos pelos de animais e que mantêm a coabitação, requerem doses mais elevadas de corticoide para controlar a doença.
Verificar se o paciente utiliza de forma correta os dispositivos de inalação para os corticoides e para os broncodilatadores: i) sincronia ao utilizar o spray; ii) uso correto do espaçador; e iii) nos dispositivos que utilizam pó, checar se o paciente é capaz de gerar um fluxo inspiratório suficiente para dispersar a droga no fluxo de ar. Dar preferência aos sistemas de liberação de medicamento que demandam fluxos inspiratórios baixos, como 20 l/min.
Descartar fatores psicossociais. Uma grande proporção de pacientes com história de resistência aos corticoides apresenta na realidade uma baixa aderência ao tratamento prescrito. Doenças como a asma demandam do paciente um esforço considerável para ser efetivamente controlada. A educação do paciente acerca de sua doença é essencial, tendo como objetivos conseguir que cooperem com o tratamento e reduzam a ansiedade diante da doença. Um plano de autotratamento deve ser elaborado e fornecido sempre por escrito. Levar em consideração fatores cognitivos como: motivação, estigma social, desatenção, distúrbios da memória, déficits associados ao envelhecimento etc.
Avaliar a presença de infecções brônquicas concomitantes, principalmente em pacientes que inalam ou ingerem altas doses de corticoides. Estes pacientes podem apresentar uma resposta imune local deficiente, predispondo a colonização de espécies oportunistas como o Mycoplasma e a Clamydia, que podem desencadear inflamação. Estes pacientes podem responder ao tratamento prolongado com macrolídeos.
Maximizar a terapêutica através de associações de drogas que facilitem a aderência e proporcionem efeito sinérgico. A combinação de corticoides com broncodilatadores de longa duração de ação é uma boa opção. Eickelberg et al.22 demonstraram que o salmeterol aumenta a translocação nuclear do GRs.
Avaliar os efeitos farmacocinéticos sistêmicos dos corticoides, objetivando avaliar se ocorre a absorção incompleta do corticoide, a conversão para a forma ativa da droga ou uma eliminação mais rápida. Pacientes com má absorção de prednisona respondem melhor com a utilização da forma líquida sob a forma de solução oral. Em pacientes com uma eliminação rápida, deve-se considerar uma segunda dose no meio da tarde.
Avaliar a presença de inflamação brônquica persistente, através de marcadores (p. ex. FeNO). Se necessário efetuar avaliações através do escarro induzido. A falência em responder aos corticoides, com persistência dos critérios de inflamação, apesar das altas doses utilizadas por inalação, ou através de teste com prednisolona oral por duas semanas, são fortes indícios para a presença de resistência aos corticoides.
Considerar a utilização de terapêutica anti-IgE, anti-IL-5/anti-IL-5R, anti-Il-4/IL-13, anti-TSLP.
Tratamento da 'Resistência aos Corticoides' na Asma
Na atualidade um importante problema para o tratamento eficaz da asma é a possibilidade de resposta reduzida aos efeitos anti-inflamatórios dos corticoides. São pacientes que apresentam asma grave com resistência ao uso do corticoide apesar de doses elevadas de corticoide inalatório ou corticoide oral.
O primeiro imunobiológico utilizado no tratamento da asma foi lançado em 2003, um anticorpo monoclonal IgG1 humanizado derivado do DNA recombinante contra IgE. A partir de 2015 surgiram os imunobiológicos anti-IL-5 e na sequência anti-IL5/R, anti-IL-4/IL-13 e anti-TSLP.
O principal objetivo terapêutico dessas drogas era o de propiciar melhor controle da doença com redução do número de exacerbações e hospitalizações e reduzir as doses de corticoide em curso (efeito poupador). Assim, no estudo MEDI-563
os indivíduos asmáticos tratados com mepolizumabe reduziram a dose de prednisona em quase 84%, em comparação com aproximadamente 48% observados naqueles no braço placebo (P = 0,04).23 No estudo ZONDA, a redução mediana na dose de corticoide oral (CO) foi de 75% para pacientes tratados com benralizumabe versus 25% com placebo.24 No estudo PONENTE que abrangeu 598 pacientes dependentes de COs tratados com benralizumabe, foi observado que mais de 80% conseguiram interromper o uso ou conseguiram reduzir a dose para 5 mg ou menos.25 Até o momento, o PONENTE destaca-se como o maior estudo poupador de CO desse gênero. Em pacientes com asma dependente de CO, o dupilumabe restringiu significativamente o seu uso em cerca de 70%, sendo que quase metade dos pacientes foi capaz de descontinuar a droga. Em paralelo ocorreram reduções nas exacerbações em 60% e melhora na função pulmonar.26
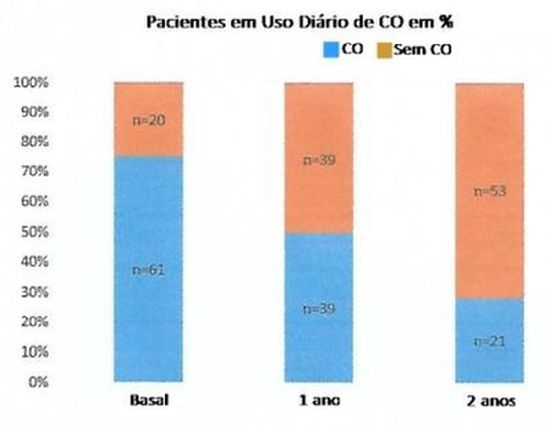
Bjerrum et al. após acompanhamento por dois anos de um grupo inicial de 81 pacientes portadores de asma grave, em que 75% utilizavam CO, tratados com mepolizumabe, reslizumabe ou benralizumabe obtiveram redução na dose de CO em 50% no primeiro ano (p < 0,001) e 28% após dois anos (p <0,001) de tratamento (Figura 1).
Figura 1 – Porcentagem de pacientes em uso diário de corticoide oral antes do tratamento anti-IL5/anti-IL-5R, após um e dois anos ou tratamento. Figura retirada com modificações de: Bjerrum AS et al. Respir Med. 2021 Jan;176:106260. ref.27
Referências
01.Barnes PJ, Adock IM. Steroid-resistent asthma. Q J Med 1995; 88:455-68.
02.Woolcock AJ. Steroid resistant asthma:what is the clinical definition? Eur Respir J 1993; 6:743-7.
03.Barnes PJ, Woolcock AJ. Difficult asthma. Eur Respir J 1998; 12:1209-18.
04.
Leung DY, Martin RJ, Szefler SJ, Sher ER, Ying S, Kay AB, Hamid Q.
Dysregulation of interleukin 4, interleukin 5, and interferon ? gene expression in steroid-resistant asthma. J Exp Med 1995; 18133-40.
05.Charmandari E, Kino T, Chrousos GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci 2004; 1024:16881.
06.Lamberts SWJ, Kioper JW, de Jong FH. Familial and iatrogenic cortisol receptor resistance. J Steroid Biochem Mol Biol 1992; 43:385-8.
07.Molnár A, Patócs A, Likó I, Nyírõ G, Rácz K, Tóth M, Sármán B. An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Medical Genetics 2018; 19:37. https://doi.org/10.1186/s12881-018-0552-6.
08.Leung DYM, Spahn JD, Szefler SJ. Immunologic bases and management of steroid-resistant asthma. Allergy Asthma Proc 1999; 20:9-14.
09.Leung DYM, Hamid Q, Vottero A et al. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor ß. J Exp Med 1997; 186:1567-74.
10.Adock IM, Brown CR, Shirasaki H, Barnes PJ. Effects of dexamethasone on cytokine and phorbol ester stimulated c-Fos and c-Jun binding and gene expression in human lung. Eur Respir J 1994; 7:2117-23.
11.Adock IM, Lane SJ, Brown CA, Lee TH, Barnes PJ. Abnormal glucocorticoid receptor/AP-1 interaction in steroid resistant asthma. J Exp Med 1995; 182:1951-8.
12.Sher ER, Leung DYM, Surs W, Kam JC, Zieg G, Kamada AK, Szefler SJ. Steroid resistant asthma. Cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest 1994; 96:33-9.
13.Adcock IM, Lane SJ Corticosteroid-insensitive asthma: molecular mechanisms. J Endocrinol 2003; 178:347-55.
14.Rhen T, Cidlowski JA. Anti-inflammatory actions of glucocorticoids: new mechanisms for old drugs. N Engl J Med 2005; 353:1711-23.
15.Hawrylowicz C, Richards D, Loke TK, Corrigan C, Lee T. A defect in corticosteroid-induced IL-10 production in T lymphocytes from corticosteroid-resistant asthmatic patients. J Allergy Clin Immunol 2002; 109:369-70.
16.Weiss ST, Lake SL, Silverman ES, Silverman EK, Richter B, Drazen JM, Tantisira KG.
Asthma steroid pharmacogenetics: a study strategy to identify replicated treatment responses. Proc Am Thorac Soc 2004; 1:364-7.
17.Stolz D, Matera MG, Rogliani P, Berge M, Papakonstantinou E, Gosens R, Singh D, Hanania N, et al. Current and future developments in the pharmacology of asthma and COPD: ERS seminar, Naples 2022. Breathe 2023; 19: 220267; DOI: 10.1183/20734735.0267-2022.
18.Al Heialy S, Ramakrishnan RK, Hamid Q. Recent advances in the immunopathogenesis of severe asthma. J Allergy Clin Immunol 2022; 149:455-465.
19.Hansbro PM, Kim RY, Starkey MR, Donovan C, Dua K, Mayall JR, Liu G, Hansbro NG, Simpson JL, Wood LG, Hirota JA, Knight DA, Foster PS, Horvat JC. Mechanisms and treatments for severe, steroid-resistant allergic airway disease and asthma. Immunol Rev 2017; 278:41-62.
20.Mei D, Tan WSD, Wong WSF. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr Opin Pharmacol 2019; 46:73-81.
21.Hirahara K, Mato N, Hagiwara K, Nakayama T. The pathogenicity of IL-33 on steroid-resistant eosinophilic inflammation via the activation of memory-type ST2+ CD4+ T cells. J Leukoc Biol 2018; 104:895-901.
22.Eickelberg O, Roth M, Lörx R, Bruce V, Rüdiger J, Johnson M, Block LH. Ligand-independent activation of the glucocorticoid receptor by beta2-adrebergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 1999; 274:1005-10.
23.Busse WW, et al. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol 2010; 125:12371244 e2.
24.Phase III ZONDA trial for benralizumab shows ability to reduce oral steroid use in severe asthma patients.Disponível na internet via WWW. Arquivo capturado em 14/07/2017. URL: https://www.astrazeneca.com/media-centre/press-releases/2017/phase-iii-zonda-trial-for-benralizumab-shows-ability-to-reduce-oral-steroid-use-in-severe-asthma-patients-22052017.
25.Menzies-Gow A, Gurnell M, Heaney LG, Corren J, Bel EH, Maspero J, Harrison T, Jackson DJ, Price D, Lugogo N, Kreindler J, Burden A, de Giorgio-Miller A, Padilla K, Martin UJ, Garcia Gil E. Oral corticosteroid elimination via a personalised reduction algorithm in adults with severe, eosinophilic asthma treated with benralizumab (PONENTE): a multicentre, open-label, single-arm study. Lancet Respir Med 2022; 10:47-58.
26. Rabe KF , Nair P , Brusselle G , Maspero JF , Castro M , Sher L , et al . Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N.Engl J Med 2018; 378: 2475 2485.
27.Bjerrum AS, Skjold T, Schmid JM. Oral corticosteroid sparing effects of anti-IL5/ anti-IL5 receptor treatment after 2 years of treatment. Respir Med 2021 Jan;176:106260.
|
|
Home
 Design by Walter Serralheiro Design by Walter Serralheiro
|
|
|